The Case Study
Accelerating Polymer Simulation with AI-Powered Coarse-Grained Modeling
How a leading materials science company achieved 100x faster simulation speed and sub-10% error—enabling new insight into complex polymer behaviors
Executive Summary
A global leader in materials science, faced a core R&D challenge: its polymer simulation workflows, though foundational to innovation, were too slow to keep up with the pace of product development. While atomistic molecular dynamics (AAMD) simulations provided the accuracy needed for evaluating polymer properties, each run could take up to 2–3 days—limiting how many material candidates could be explored in a development cycle. Faster coarse-grained (CGMD) methods existed – a class of simulations that simplify molecular detail to accelerate runtime –but leading approaches like Martini 3 were originally designed for biomolecular systems — not polymers – resulting in significant accuracy limitations when applied to polymer simulation.
To overcome this, the materials science company partnered with Multiscale Technologies and deployed the MIND platform—Multiscale’s enterprise product for model development and AIpowered optimization of simulation workflows. Using MIND, Multiscale implemented a Multi-Objective Bayesian Optimization (MOBO) framework to refine CGMD models, dramatically improving accuracy while accelerating simulation time.
The result: simulations that were 100x faster while maintaining <10% error compared to atomistic benchmarks. Beyond speed, the science company gained access to new levels of mesoscopic insight—critical for advanced material research. The project validated the use of AI-enhanced CGMD at scale and established a foundation for broader adoption across the company’s R&D pipeline, including co-polymer design and simulation-driven discovery.
Business Problem
Polymer simulation is a cornerstone of the company’s R&D efforts. The company’s simulation team routinely performs atomistic molecular dynamics (AAMD) to evaluate the physical properties of polymer candidates. While accurate, these simulations are time- and resource-intensive—each configuration can take 2–3 days to complete. This limited how many candidates could be explored in a given R&D cycle. This created a major bottleneck. Although coarse-grained molecular dynamics (CGMD) methods like Martini 3 offered a faster alternative, they were designed for biomolecules such as proteins and lipids – not polymers – resulting in poor accuracy for polymer property predictions and limiting their use in high confidence decisions. The company needed a new approach that could match the fidelity of AAMD but deliverresults at CGMD speeds.
Simulation Bottlenecks in Existing Workflows:
- Atomistic simulations were accurate but slow, often taking 2–3 days per system.
- Coarse-grained models like Martini3 promised faster simulations, but their design for biomolecules limited their accuracy and usability for polymer systems.
- This dual limitation constrained both speed and insight in R&D workflows.
Technical Challenge
The materials science company needed to simulate complex polymer systems efficiently while maintaining high fidelity in key material property predictions.
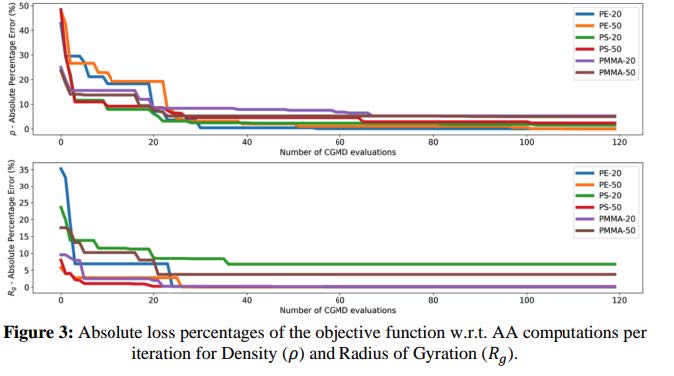
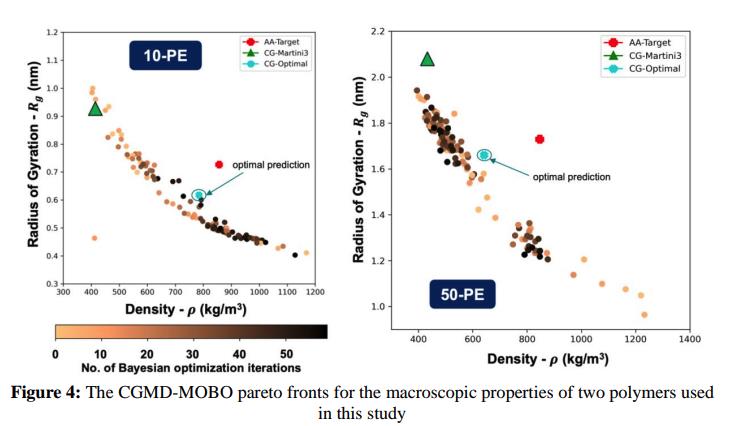
The primary challenge was accuracy: Baseline Martini 3 CG models, while effective for biomolecular systems, introduced significant errors when applied to polymers resulting in up to 60% deviation in key properties like density (ρ) and radius of gyration (Rg) compared to atomistic benchmarks. These inaccuracies prevented the company from using CGMD to simulate polymer systems effectively.
Moreover, the traditional CGMD setup could not adapt well to polymers with varying degrees of polymerization, further limiting its utility for iterative materials development.
Multiscale’s Solution
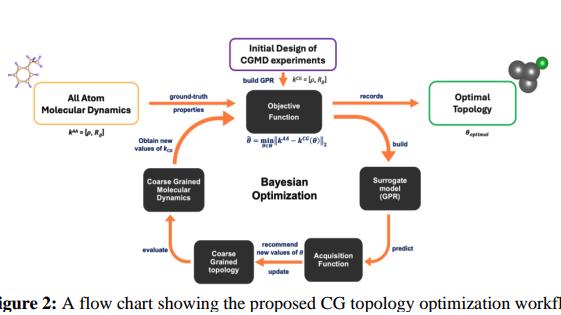
To address the dual challenge of speed and accuracy in polymer simulations, the company partnered with Multiscale Technologies and leveraged the MIND platform —Multiscale’s enterprise product for model development and simulation workflow optimization. The platform provided the infrastructure to integrate domain-specific molecular dynamics (MD) workflows with scalable, AI-powered optimization. Rather than relying on out-of-the-box CGMD models, which were not designed for polymer systems, the team used MIND to systematically re-parameterize molecular topologies for polymer-specific accuracy.
Multiscale used the MIND platform to design and run a Multi-Objective Bayesian Optimization (MOBO) scheme that automatically refined coarse-grained molecular topologies while targeting high-fidelity outputs. The platform handled the full learning loop—from data ingestion and topology transformation to AI-driven parameter search and model convergence. The system was configured for rapid iteration and robust transferability, enabling the company team to evaluate and adapt the solution with minimal dependency on external code.
The materials science company’s simulation experts provided domain guidance on the molecular systems and validation methods. This collaborative setup ensured scientific rigor, operational independence, and seamless integration into company’s internal toolchain.
Key Outcomes at a Glance
100x
Simulation speedup compared to AAMD.
<10%
Deviation from AAMD reference values in both density and Rg.<10%
Errors with Multiscale optimization – improvement from 60% error (Martini 3).~50
Iterations per polymer system achieved convergence.“What used to take us two to three days per simulation, we can now do in under an hour. That kind of speedup fundamentally changes how we approach polymer screening.”
Lead Simulation Engineer
“Beyond speed, the real breakthrough is that we’re now simulating mesoscopic behaviors—like phase separation—that we simply couldn’t capture with traditional atomistic models.”
Senior Materials Researcher
“This project proved that AI-enhanced coarse-grained modeling isn’t just viable—it’s a strategic enabler for the future of polymer design.”
Director of R&D Strategy
Business Outcomes
This collaboration served as a strategic validation of AI-powered simulation workflows in high-stakes R&D environments. By reducing simulation time by 100x while preserving fidelity, the company significantly increased throughput in its polymer development efforts. The team can now explore broader design spaces and iterate more frequently—without compromising scientific rigor.
The project also laid the foundation for continued innovation. The company is now planning to extend the AI-optimized CGMD framework to model co-polymer compositions, with the potential to scale adoption across its 100-person R&D modeling team. This foundational work is expected to support broader simulation-driven design initiatives in polymer science.